After smelling so much of E. coli culture (which pretty resembles our pee)...
We are at the final stage! Harvesting.
This stage involves the isolation and purification of product.
Isolation
Isolation basically means setting our product apart from the rest of the culture broth. But before doing so, we need to first look at the nature of our product.
In this case, our product of interest is Green Fluorescent Protein (GFP) – an intracellular product (produced within the cell). Therefore, it is necessary to lyse the bacteria cells to release those proteins. Evil humans, aren’t we? We have to kill those E. coli for GFP after treating them so well.
 Well, well. Illustrations are indeed BO-RING. So here’s a picture we took:
Well, well. Illustrations are indeed BO-RING. So here’s a picture we took:
*TADA* Have a look at our tubes under the UV light.
First tube from left contains the supernatant (liquid broth) separated from cell pellet in tube 2 after centrifugation.
Second tube contains only the bacteria cell pellet. Notice how the pellet fluoresces under the UV light.
Third tube is the tube containing both supernatant and pellet.
Without UV light, it looks like this:
 And now, with our bacteria cells, it’s time to kill them! *Evil laughters*
And now, with our bacteria cells, it’s time to kill them! *Evil laughters*I seriously think that we are some sort of mad scientists.
To do that, three methods of cell disruption (cell torturing?) are performed.
- Using enzymes (Chomping their membranes apart?)
- Freezing and Thawing (To induce Temperature shock)
- Sonication (Ultrasonic waves!)
Method 1: Using Enzymes
Firstly, we resuspend the pellet in 500µL of TE buffer of pH 7.5 using a micropipettor until there are no visible clumps. Then, we added two drops of lysozyme to the resuspended cell pellet – this initiates the enzymatic digestion of the E. coli cell wall.
This will initiate the enzymatic digestion of the bacteria cell wall. Allow the enzymes to act for 15 minutes. Such a poor thing that they have to die so slowly...
 à Resuspended pellet in TAE buffer and lysozyme.
à Resuspended pellet in TAE buffer and lysozyme.
Method 2: Freezing and Thawing
I really love this part as it involves liquid nitrogen!
After enzymatic digestion, our next ‘torturing’ phrase involved this particular liquid that is of -196°C (Very very cold). Even though it looks pretty like dry ice (Cos they give off lovely vapours too), but they’re pretty dangerous. So never place your hand inside liquid nitrogen!

The diagram shows the process of freezing with liquid nitrogen. Yup it looks pretty much like water. I could still remember hearing those sizzling sounds and seeing those vapors as we dip our tubes inside it.
Method 3: Sonication
Our last torturing phase involves the use of ultrasonic waves which completes the cell disruption. During the process, ear muffs are required because those waves are of higher frequency than sound. Sonication causes bacteria cell wall to implode under the vibrational pressure. It is done on ice for 4 cycles of 25 seconds with 10 seconds rest in between Sonication cycles.

And here’s Ah Guan and Hafiz who did the sonications. Don’t they look like DJs at work? Cool ear muffs - Kinda stylo, don’t you think?


After cell disruption, we spun the contents of the tube again in the centrifuge for 20 minutes at 10,000 rpm. Then, like what was done previously, the pellet and supernatant were visualized under the UV light again.
 *Notice that the supernatant is fluorescing instead of the pellet now. With the cells being ruptured, green fluorescence proteins escaped the boundaries of the cell and form one of the supernatant’s constituent. The pellet constitutes the cell debris which does not glow due to the absence of GFP.
*Notice that the supernatant is fluorescing instead of the pellet now. With the cells being ruptured, green fluorescence proteins escaped the boundaries of the cell and form one of the supernatant’s constituent. The pellet constitutes the cell debris which does not glow due to the absence of GFP.Purification
With the extract obtained from isolation earlier, it’s time to purify them!
Purification means the removal of impurities – cell debris especially after lysing of cells.
It is necessary to purify our extract so that the product can be used for other purposes. In this experiment, we used gel filtration or gel permeation chromatography to purify our GFP product. Such method of purification uses a column of a polymer gel resins (Sephadex G75). The resins contain very small pores in which molecules that are small enough can diffuse within. Hence, when the extract is poured into the column, the larger molecules will flow through the column faster where the smaller molecules will spend more time interacting and diffusing into the pores of gel resins. This achieves separation of different molecules by size.
Below shows an illustration of how the column looks like:
 This looks pretty complex. But in actual fact, the column looks like that:
This looks pretty complex. But in actual fact, the column looks like that: The resins are small and transparent.
The resins are small and transparent.Note the difference in buffer layer and the layer containing the gel matrix.
Procedure of purification of GFP:
We have labeled 9 tubes (“1 to 8” and “blank”) with a mark at 2ml level. The column was carefully drained into the waste breaker to adjust the buffer level such that it is just even with the top of the gel bed. Cell-free extract was then transferred to the top of the gel bed by gently stirring the pipet around the inside edge of the column, just above the top of the packed matrix. (Extra care is taken to not disturb the matrix)
We then start taking fractions by placing one labeled tube (tube 1) under the stopcock, and carefully releasing the stopcock to collect the eluant (buffer). The flow rate was adjusted to a 1drop/2 sec interval. Precautions are taken to not let the entire column run dry by continuously adding 50mM ammonium bicarbonate buffer to the top of the column when fractions are taken.
The same collecting procedures were done till the all 8 tubes were filled.
With all samples collected, we then transfered them to plastic cuvettes for measuring of absorbance. (This part done by our dear Miss 3!)
Before measuring of absorbance, we must always first have our blank to zero our readings. In this case, we used ammonium sulphate buffer (without any sample) as our blank.

More pics of purification and sonication. Credits to Miss LoanShark
Result for Isolation and Purification of GFP
(By Miss 3)
.bmp)

Answers to Questions:
1. Based on the graph, the peak occurred at fraction 2. In general, the higher the concentration of absorbing substance, which in this case, green fluorescent protein (GFP) in a sample, the greater the amount of light will be absorbed i.e. a high absorbance reading will be reflected. Since fraction 2 had the highest absorbance, this would mean that most of our GFP was collected in this tube.
The absorbance for the first fraction will usually be low due to the fact that when the sample was first added to the chromatography column, it takes some time for it to reach the other end of the column. Hence, what was collected in fraction 1 will be mostly the solution that was contained previously in the column i.e. ammonium bicarbonate. From fraction 2 onwards, the GFP will start to be eluted. Hence, the absorbance will be observed to increase till it reached a peak. Once it had reached a peak, it would mean that most of the GFP have been eluted. Therefore, the subsequent fraction will have lesser GFP contained in it, and the absorbance will be observed to decrease significantly.
2. For size exclusion chromatography, as the proteins travel down the column, the low molecular weight proteins will interact and diffuse into the pores of the gel resins, retarding their flow. On the other hand, the higher molecular weight proteins are less able to enter the pores of the gel resins, so they will be eluted first. Therefore in this case, a protein with a Mr of 50,000 kD will elute in a fraction before GFP, which has a Mr of 27,000 kD.
Guess that ends what I have to say for what we did during isolation and purification of our product.
MissET aka LaoDa ;)


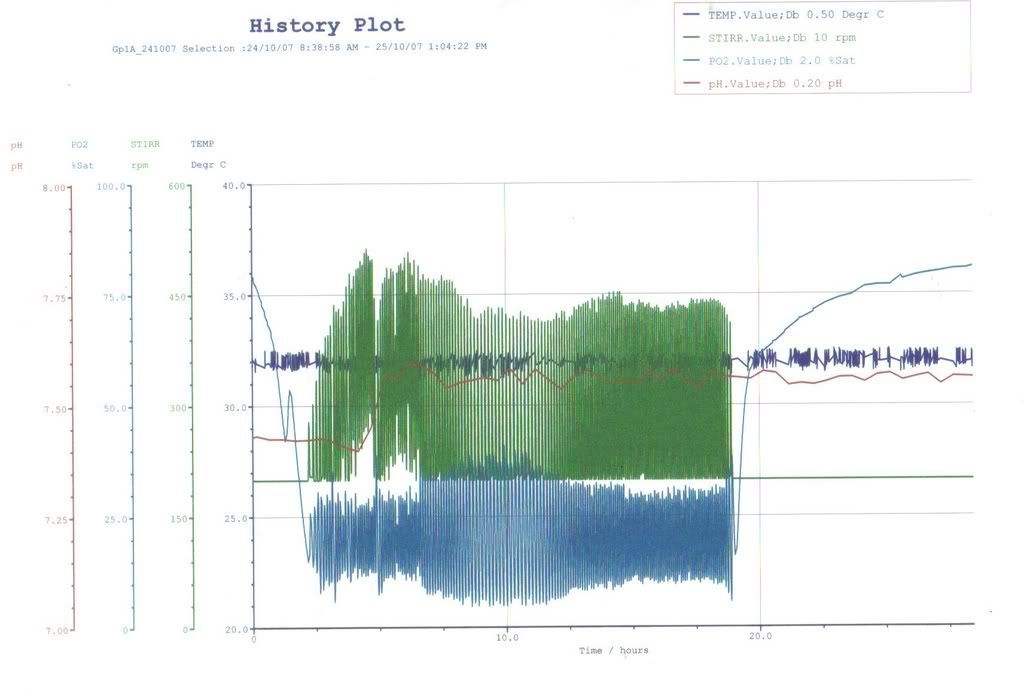
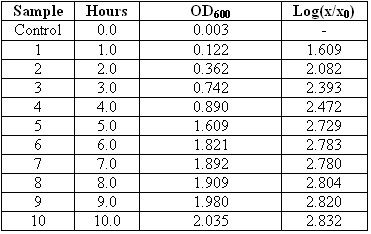
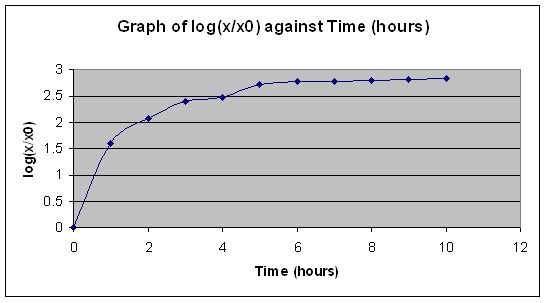


 Ok… and as for preparing the fermenter itself…we didn’t really do much…it was more like..the teacher and the DT doing it..so I ain’t gonna copy and paste la hor.. XD
Ok… and as for preparing the fermenter itself…we didn’t really do much…it was more like..the teacher and the DT doing it..so I ain’t gonna copy and paste la hor.. XD
.bmp)








